

A Certain Chemical Reaction Releases

A thermite reaction using iron(Iii) oxide. The sparks flying outwards are globules of molten iron abaft smoke in their wake.
A chemical reaction is a process that leads to the chemical transformation of i set of chemical substances to some other.[1] Classically, chemical reactions embrace changes that only involve the positions of electrons in the forming and breaking of chemical bonds between atoms, with no modify to the nuclei (no change to the elements nowadays), and can oft be described by a chemical equation. Nuclear chemistry is a sub-field of study of chemistry that involves the chemic reactions of unstable and radioactive elements where both electronic and nuclear changes can occur.
The substance (or substances) initially involved in a chemical reaction are called reactants or reagents. Chemical reactions are usually characterized by a chemic change, and they yield one or more products, which usually have properties unlike from the reactants. Reactions frequently consist of a sequence of private sub-steps, the and then-chosen unproblematic reactions, and the data on the precise form of activeness is function of the reaction machinery. Chemical reactions are described with chemical equations, which symbolically nowadays the starting materials, end products, and sometimes intermediate products and reaction conditions.
Chemical reactions happen at a characteristic reaction charge per unit at a given temperature and chemical concentration. Typically, reaction rates increase with increasing temperature because there is more thermal energy available to achieve the activation energy necessary for breaking bonds between atoms. Reactions may proceed in the frontward or reverse direction until they get to completion or accomplish equilibrium. Reactions that keep in the forwards direction to arroyo equilibrium are often described as spontaneous and reduce the free energy if they occur at abiding temperature and force per unit area. Non-spontaneous reactions require input of free energy to go forward (examples include charging a battery driven past an external electric power source, or photosynthesis driven by absorption of electromagnetic radiation usually in the grade of sunlight).
A reaction may be classified as redox in which oxidation and reduction occur or nonredox in which there is no oxidation and reduction occurring. About simple redox reactions may be classified as combination, decomposition, or single displacement reactions.
Different chemical reactions are used during chemical synthesis in order to obtain a desired product. In biochemistry, a consecutive serial of chemical reactions (where the product of ane reaction is the reactant of the next reaction) class metabolic pathways. These reactions are ofttimes catalyzed by protein enzymes. Enzymes increase the rates of biochemical reactions, so that metabolic syntheses and decompositions impossible under ordinary conditions tin can occur at the temperature and concentrations nowadays inside a cell.
The full general concept of a chemic reaction has been extended to reactions between entities smaller than atoms, including nuclear reactions, radioactive decays, and reactions between simple particles, as described past quantum field theory.
History

Antoine Lavoisier developed the theory of combustion as a chemic reaction with oxygen.
Chemical reactions such as combustion in fire, fermentation and the reduction of ores to metals were known since artifact. Initial theories of transformation of materials were developed by Greek philosophers, such as the 4-Element Theory of Empedocles stating that any substance is composed of the 4 basic elements – fire, water, air and earth. In the Middle Ages, chemical transformations were studied past alchemists. They attempted, in item, to convert lead into golden, for which purpose they used reactions of pb and lead-copper alloys with sulfur.[two]
The artificial product of chemical substances already was a primal goal for medieval alchemists.[3] Examples include the synthesis of ammonium chloride from organic substances as described in the works (c. 850–950) attributed to Jābir ibn Ḥayyān,[iv] or the production of mineral acids such as sulfuric and nitric acids past later alchemists, starting from c. 1300.[5] The production of mineral acids involved the heating of sulfate and nitrate minerals such equally copper sulfate, alum and saltpeter. In the 17th century, Johann Rudolph Glauber produced hydrochloric acid and sodium sulfate by reacting sulfuric acid and sodium chloride. With the development of the lead chamber process in 1746 and the Leblanc procedure, allowing large-calibration production of sulfuric acrid and sodium carbonate, respectively, chemical reactions became implemented into the manufacture. Further optimization of sulfuric acrid technology resulted in the contact process in the 1880s,[6] and the Haber process was developed in 1909–1910 for ammonia synthesis.[7]
From the 16th century, researchers including January Baptist van Helmont, Robert Boyle, and Isaac Newton tried to constitute theories of the experimentally observed chemical transformations. The phlogiston theory was proposed in 1667 by Johann Joachim Becher. It postulated the being of a burn down-similar element called "phlogiston", which was independent within flammable bodies and released during combustion. This proved to be false in 1785 by Antoine Lavoisier who found the correct caption of the combustion every bit reaction with oxygen from the air.[viii]
Joseph Louis Gay-Lussac recognized in 1808 that gases e'er react in a sure relationship with each other. Based on this idea and the atomic theory of John Dalton, Joseph Proust had developed the police of definite proportions, which later resulted in the concepts of stoichiometry and chemic equations.[9]
Regarding the organic chemistry, it was long believed that compounds obtained from living organisms were likewise complex to exist obtained synthetically. According to the concept of vitalism, organic thing was endowed with a "vital force" and distinguished from inorganic materials. This separation was ended nevertheless by the synthesis of urea from inorganic precursors by Friedrich Wöhler in 1828. Other chemists who brought major contributions to organic chemical science include Alexander William Williamson with his synthesis of ethers and Christopher Kelk Ingold, who, among many discoveries, established the mechanisms of substitution reactions.
Characteristics
| | This section needs expansion. You tin help by calculation to it. (Nov 2020) |
The general characteristics of chemical reactions are:
- Evolution of a gas
- Germination of a precipitate
- Change in temperature
- Alter in country
Equations
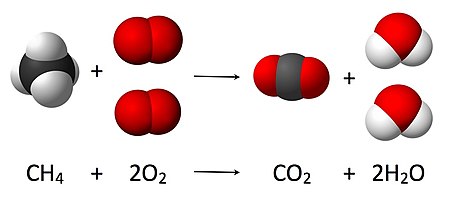
Equally seen from the equation CHiv + 2O2 → CO2 + 2 H2O, a coefficient of 2 must be placed before the oxygen gas on the reactants side and before the water on the products side in society for, as per the law of conservation of mass, the quantity of each chemical element does not alter during the reaction
Chemical equations are used to graphically illustrate chemic reactions. They consist of chemical or structural formulas of the reactants on the left and those of the products on the right. They are separated past an arrow (→) which indicates the management and type of the reaction; the pointer is read as the give-and-take "yields".[10] The tip of the arrow points in the direction in which the reaction proceeds. A double pointer (⇌) pointing in opposite directions is used for equilibrium reactions. Equations should be balanced according to the stoichiometry, the number of atoms of each species should be the aforementioned on both sides of the equation. This is achieved by scaling the number of involved molecules (A, B, C and D in a schematic case below) by the appropriate integers a, b, c and d.[11]
- a A + b B → c C + d D
More than elaborate reactions are represented past reaction schemes, which in add-on to starting materials and products show important intermediates or transition states. Also, some relatively minor additions to the reaction tin can exist indicated above the reaction arrow; examples of such additions are h2o, heat, illumination, a catalyst, etc. Similarly, some modest products can be placed below the arrow, often with a minus sign.
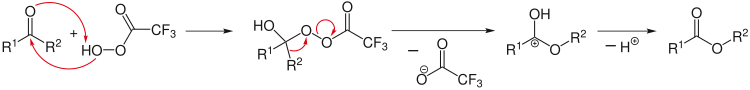
Retrosynthetic analysis can exist applied to design a complex synthesis reaction. Here the analysis starts from the products, for example past splitting selected chemic bonds, to arrive at plausible initial reagents. A special arrow (⇒) is used in retro reactions.[12]
Elementary reactions
The uncomplicated reaction is the smallest segmentation into which a chemic reaction can be decomposed, it has no intermediate products.[xiii] About experimentally observed reactions are built up from many uncomplicated reactions that occur in parallel or sequentially. The actual sequence of the private elementary reactions is known as reaction mechanism. An unproblematic reaction involves a few molecules, usually one or two, because of the depression probability for several molecules to meet at a certain time.[14]

Isomerization of azobenzene, induced by calorie-free (hν) or heat (Δ)
The most important uncomplicated reactions are unimolecular and bimolecular reactions. Only one molecule is involved in a unimolecular reaction; it is transformed past an isomerization or a dissociation into one or more other molecules. Such reactions require the addition of energy in the form of oestrus or light. A typical example of a unimolecular reaction is the cis–trans isomerization, in which the cis-course of a compound converts to the trans-form or vice versa.[15]
In a typical dissociation reaction, a bond in a molecule splits (ruptures) resulting in 2 molecular fragments. The splitting tin be homolytic or heterolytic. In the commencement case, the bond is divided so that each production retains an electron and becomes a neutral radical. In the second case, both electrons of the chemical bond remain with i of the products, resulting in charged ions. Dissociation plays an of import role in triggering chain reactions, such as hydrogen–oxygen or polymerization reactions.
- Dissociation of a molecule AB into fragments A and B

For bimolecular reactions, two molecules collide and react with each other. Their merger is called chemic synthesis or an improver reaction.

Another possibility is that merely a portion of one molecule is transferred to the other molecule. This type of reaction occurs, for instance, in redox and acid–base reactions. In redox reactions, the transferred particle is an electron, whereas in acid–base reactions it is a proton. This type of reaction is also called metathesis.

for example

Chemical equilibrium
Most chemic reactions are reversible; that is, they can and practise run in both directions. The forward and reverse reactions are competing with each other and differ in reaction rates. These rates depend on the concentration and therefore alter with time of the reaction: the reverse rate gradually increases and becomes equal to the rate of the forward reaction, establishing the then-called chemical equilibrium. The time to reach equilibrium depends on parameters such as temperature, pressure, and the materials involved, and is determined past the minimum gratis energy. In equilibrium, the Gibbs energy must be naught. The pressure dependence can exist explained with the Le Chatelier'southward principle. For example, an increase in pressure due to decreasing volume causes the reaction to shift to the side with the fewer moles of gas.[16]
The reaction yield stabilizes at equilibrium, but tin can be increased past removing the production from the reaction mixture or inverse by increasing the temperature or force per unit area. A change in the concentrations of the reactants does non affect the equilibrium constant, but does affect the equilibrium position.
Thermodynamics
Chemical reactions are determined by the laws of thermodynamics. Reactions can proceed by themselves if they are exergonic, that is if they release free energy. The associated costless energy alter of the reaction is composed of the changes of two dissimilar thermodynamic quantities, enthalpy and entropy:[17]
-
- .
- Yard: free energy, H: enthalpy, T: temperature, S: entropy, Δ: deviation (change between original and production)

Reactions can be exothermic, where ΔH is negative and energy is released. Typical examples of exothermic reactions are combustion, precipitation and crystallization, in which ordered solids are formed from matted gaseous or liquid phases. In contrast, in endothermic reactions, heat is consumed from the environment. This can occur by increasing the entropy of the system, often through the formation of gaseous or dissolved reaction products, which have higher entropy. Since the entropy term in the costless-free energy change increases with temperature, many endothermic reactions preferably take identify at loftier temperatures. On the reverse, many exothermic reactions such as crystallization occur preferably at lower temperatures. A modify in temperature can sometimes reverse the sign of the enthalpy of a reaction, as for the carbon monoxide reduction of molybdenum dioxide:
- ;


This reaction to grade carbon dioxide and molybdenum is endothermic at low temperatures, condign less so with increasing temperature.[18] ΔH° is zero at 1855 K, and the reaction becomes exothermic in a higher place that temperature.
Changes in temperature tin also opposite the direction trend of a reaction. For example, the h2o gas shift reaction

is favored by low temperatures, but its reverse is favored by high temperature. The shift in reaction management tendency occurs at 1100 K.[18]
Reactions can also be characterized past their internal energy alter, which takes into business relationship changes in the entropy, book and chemical potentials. The latter depend, among other things, on the activities of the involved substances.[19]
-
- U: internal free energy, S: entropy, p: force per unit area, μ: chemic potential, n: number of molecules, d: small-scale modify sign

Kinetics
The speed at which reactions takes place is studied by reaction kinetics. The rate depends on diverse parameters, such as:
- Reactant concentrations, which usually make the reaction happen at a faster charge per unit if raised through increased collisions per unit time. Some reactions, however, have rates that are independent of reactant concentrations, due to a express number of catalytic sites. These are chosen zero order reactions.
- Expanse available for contact between the reactants, in particular solid ones in heterogeneous systems. Larger surface areas atomic number 82 to higher reaction rates.
- Pressure – increasing the pressure decreases the volume between molecules and therefore increases the frequency of collisions between the molecules.
- Activation free energy, which is defined as the amount of energy required to make the reaction beginning and acquit on spontaneously. Higher activation energy implies that the reactants need more than energy to start than a reaction with a lower activation energy.
- Temperature, which hastens reactions if raised, since higher temperature increases the energy of the molecules, creating more than collisions per unit time,
- The presence or absence of a catalyst. Catalysts are substances which brand weak bonds with reactants or intermediates and change the pathway (mechanism) of a reaction which in turn increases the speed of a reaction past lowering the activation energy needed for the reaction to take place. A catalyst is not destroyed or changed during a reaction, then information technology can be used over again.
- For some reactions, the presence of electromagnetic radiations, virtually notably ultraviolet light, is needed to promote the breaking of bonds to start the reaction. This is especially true for reactions involving radicals.
Several theories permit computing the reaction rates at the molecular level. This field is referred to as reaction dynamics. The rate v of a get-go-society reaction, which could exist disintegration of a substance A, is given by:
![{\displaystyle v=-{\frac {d[{\ce {A}}]}{dt}}=k\cdot [{\ce {A}}].}](https://wikimedia.org/api/rest_v1/media/math/render/svg/12291760fcaff20a02ff74abd0dfcb922664cddb)
Its integration yields:
![{\displaystyle {\ce {[A]}}(t)={\ce {[A]}}_{0}\cdot e^{-k\cdot t}.}](https://wikimedia.org/api/rest_v1/media/math/render/svg/498c37558508e2f7297604f93bb5408dcd8c3fd4)
Here k is the kickoff-order rate abiding, having dimension 1/time, [A](t) is concentration at a time t and [A]0 is the initial concentration. The rate of a first-order reaction depends only on the concentration and the properties of the involved substance, and the reaction itself tin can be described with a characteristic one-half-life. More than one time constant is needed when describing reactions of college order. The temperature dependence of the rate abiding ordinarily follows the Arrhenius equation:

where E a is the activation free energy and k B is the Boltzmann constant. 1 of the simplest models of reaction rate is the standoff theory. More than realistic models are tailored to a specific problem and include the transition state theory, the calculation of the potential energy surface, the Marcus theory and the Rice–Ramsperger–Kassel–Marcus (RRKM) theory.[20]
Reaction types
Four basic types
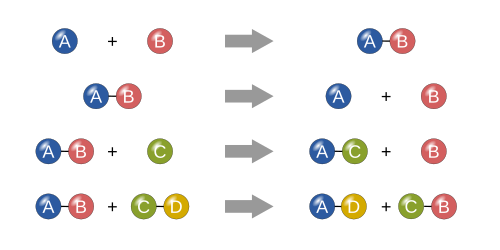
Representation of four basic chemical reactions types: synthesis, decomposition, single replacement and double replacement.
Synthesis
In a synthesis reaction, 2 or more than uncomplicated substances combine to form a more complex substance. These reactions are in the general form:

2 or more than reactants yielding one product is some other way to identify a synthesis reaction. One example of a synthesis reaction is the combination of iron and sulfur to form atomic number 26(II) sulfide:

Another instance is unproblematic hydrogen gas combined with simple oxygen gas to produce a more complex substance, such as water.[21]
Decomposition
A decomposition reaction is when a more complex substance breaks down into its more elementary parts. It is thus the opposite of a synthesis reaction, and tin be written as[21] [22]

One instance of a decomposition reaction is the electrolysis of water to make oxygen and hydrogen gas:

Single displacement
In a unmarried deportation reaction, a single uncombined element replaces some other in a compound; in other words, ane element trades places with another element in a compound[21] These reactions come in the full general form of:

One example of a single displacement reaction is when magnesium replaces hydrogen in h2o to make magnesium hydroxide and hydrogen gas:

Double displacement
In a double displacement reaction, the anions and cations of ii compounds switch places and form two entirely different compounds.[21] These reactions are in the general form:[22]

For example, when barium chloride (BaCltwo) and magnesium sulfate (MgSO4) react, the SO4 2− anion switches places with the 2Cl− anion, giving the compounds BaSO4 and MgCl2.
Another example of a double displacement reaction is the reaction of lead(Two) nitrate with potassium iodide to form lead(II) iodide and potassium nitrate:

Combustion
In a combustion reaction, an element or compound reacts with an oxidant, usually oxygen, often producing energy in the course of oestrus or low-cal. Combustion reactions frequently involve a hydrocarbon. For instance, combustion of i mole (114 g) of octane in oxygen

releases 5500 kJ. A combustion reaction can also event from carbon, magnesium or sulfur reacting with oxygen.[23]


Oxidation and reduction

Analogy of a redox reaction

Sodium chloride is formed through the redox reaction of sodium metal and chlorine gas
Redox reactions can exist understood in terms of transfer of electrons from i involved species (reducing agent) to another (oxidizing amanuensis). In this process, the former species is oxidized and the latter is reduced. Though sufficient for many purposes, these descriptions are not precisely right. Oxidation is better defined as an increase in oxidation state of atoms, and reduction as a decrease in oxidation state. In practice, the transfer of electrons will always alter the oxidation state, but there are many reactions that are classed every bit "redox" even though no electron transfer occurs (such equally those involving covalent bonds).[24] [25]
In the post-obit redox reaction, hazardous sodium metallic reacts with toxic chlorine gas to form the ionic chemical compound sodium chloride, or mutual table salt:

In the reaction, sodium metal goes from an oxidation state of 0 (as information technology is a pure chemical element) to +1: in other words, the sodium lost 1 electron and is said to accept been oxidized. On the other manus, the chlorine gas goes from an oxidation of 0 (information technology is also a pure element) to −1: the chlorine gains i electron and is said to accept been reduced. Because the chlorine is the ane reduced, it is considered the electron acceptor, or in other words, induces oxidation in the sodium – thus the chlorine gas is considered the oxidizing agent. Conversely, the sodium is oxidized or is the electron donor, and thus induces reduction in the other species and is considered the reducing agent.
Which of the involved reactants would exist reducing or oxidizing agent can be predicted from the electronegativity of their elements. Elements with low electronegativity, such as near metals, easily donate electrons and oxidize – they are reducing agents. On the contrary, many oxides or ions with loftier oxidation numbers of their non-oxygen atoms, such as H
ii O
2 , MnO −
4 , CrO
three , Cr
two O 2−
7 , or OsO
4 , can proceeds 1 or two extra electrons and are strong oxidizing agents.
For some principal-group elements the number of electrons donated or accustomed in a redox reaction can be predicted from the electron configuration of the reactant element. Elements try to reach the low-free energy element of group 0 configuration, and therefore alkali metals and halogens will donate and take i electron, respectively. Noble gases themselves are chemically inactive.[26]
The overall redox reaction can be balanced by combining the oxidation and reduction half-reactions multiplied by coefficients such that the number of electrons lost in the oxidation equals the number of electrons gained in the reduction.
An important class of redox reactions are the electrolytic electrochemical reactions, where electrons from the power supply at the negative electrode are used as the reducing agent, and electron withdrawal at the positive electrode as the oxidizing amanuensis. These reactions are specially important for the production of chemical elements, such every bit chlorine[27] or aluminium. The reverse process, in which electrons are released in redox reactions and chemical energy is converted to electric energy, is possible and used in batteries.
Complexation
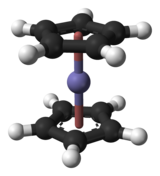
In complexation reactions, several ligands react with a metal atom to form a coordination circuitous. This is accomplished by providing lone pairs of the ligand into empty orbitals of the metal atom and forming dipolar bonds. The ligands are Lewis bases, they tin be both ions and neutral molecules, such as carbon monoxide, ammonia or h2o. The number of ligands that react with a central metal cantlet tin can be constitute using the 18-electron rule, saying that the valence shells of a transition metal will collectively accommodate 18 electrons, whereas the symmetry of the resulting complex can exist predicted with the crystal field theory and ligand field theory. Complexation reactions also include ligand exchange, in which 1 or more than ligands are replaced by another, and redox processes which alter the oxidation land of the central metal atom.[28]
Acid–base reactions
In the Brønsted–Lowry acid–base theory, an acrid–base reaction involves a transfer of protons (H+) from one species (the acid) to some other (the base). When a proton is removed from an acid, the resulting species is termed that acid's conjugate base. When the proton is accepted by a base, the resulting species is termed that base of operations's cohabit acrid.[29] In other words, acids act as proton donors and bases human action as proton acceptors according to the following equation:

The reverse reaction is possible, and thus the acid/base of operations and conjugated base/acrid are always in equilibrium. The equilibrium is adamant by the acid and base dissociation constants (K a and G b) of the involved substances. A special case of the acid–base reaction is the neutralization where an acid and a base of operations, taken at exactly aforementioned amounts, grade a neutral common salt.
Acid–base reactions can have dissimilar definitions depending on the acid–base concept employed. Some of the nigh common are:
- Arrhenius definition: Acids dissociate in water releasing HthreeO+ ions; bases dissociate in water releasing OH− ions.
- Brønsted–Lowry definition: Acids are proton (H+) donors, bases are proton acceptors; this includes the Arrhenius definition.
- Lewis definition: Acids are electron-pair acceptors, bases are electron-pair donors; this includes the Brønsted-Lowry definition.
Precipitation

Precipitation is the formation of a solid in a solution or inside another solid during a chemical reaction. It usually takes place when the concentration of dissolved ions exceeds the solubility limit[thirty] and forms an insoluble common salt. This process can be assisted by adding a precipitating agent or by removal of the solvent. Rapid precipitation results in an amorphous or microcrystalline residue and wearisome process tin yield single crystals. The latter tin as well be obtained past recrystallization from microcrystalline salts.[31]
Solid-state reactions
Reactions can take place betwixt 2 solids. However, because of the relatively small diffusion rates in solids, the corresponding chemical reactions are very slow in comparison to liquid and gas stage reactions. They are accelerated by increasing the reaction temperature and finely dividing the reactant to increase the contacting surface area.[32]
Reactions at the solid/gas interface
Reaction tin take place at the solid|gas interface, surfaces at very depression pressure level such every bit ultra-loftier vacuum. Via scanning tunneling microscopy, information technology is possible to find reactions at the solid|gas interface in real infinite, if the time scale of the reaction is in the correct range.[33] [34] Reactions at the solid|gas interface are in some cases related to catalysis.
Photochemical reactions
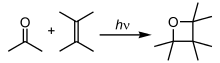
In photochemical reactions, atoms and molecules blot energy (photons) of the illumination light and convert into an excited state. They can then release this free energy by breaking chemic bonds, thereby producing radicals. Photochemical reactions include hydrogen–oxygen reactions, radical polymerization, chain reactions and rearrangement reactions.[35]
Many important processes involve photochemistry. The premier example is photosynthesis, in which most plants employ solar free energy to convert carbon dioxide and water into glucose, disposing of oxygen every bit a side-product. Humans rely on photochemistry for the formation of vitamin D, and vision is initiated by a photochemical reaction of rhodopsin.[15] In fireflies, an enzyme in the belly catalyzes a reaction that results in bioluminescence.[36] Many pregnant photochemical reactions, such equally ozone germination, occur in the Earth atmosphere and constitute atmospheric chemical science.
Catalysis

Schematic potential free energy diagram showing the issue of a catalyst in an endothermic chemical reaction. The presence of a catalyst opens a dissimilar reaction pathway (in red) with a lower activation energy. The terminal consequence and the overall thermodynamics are the same.

Solid heterogeneous catalysts are plated on meshes in ceramic catalytic converters in order to maximize their expanse. This frazzle converter is from a Peugeot 106 S2 1100
In catalysis, the reaction does not proceed straight, but through reaction with a third substance known every bit catalyst. Although the catalyst takes part in the reaction, forming weak bonds with reactants or intermediates, it is returned to its original state by the terminate of the reaction then is not consumed. However, it can exist inhibited, deactivated or destroyed by secondary processes. Catalysts can be used in a dissimilar stage (heterogeneous) or in the same stage (homogeneous) as the reactants. In heterogeneous catalysis, typical secondary processes include coking where the catalyst becomes covered by polymeric side products. Additionally, heterogeneous catalysts can dissolve into the solution in a solid–liquid system or evaporate in a solid–gas system. Catalysts can only speed up the reaction – chemicals that slow downwards the reaction are called inhibitors.[37] [38] Substances that increment the action of catalysts are called promoters, and substances that deactivate catalysts are chosen catalytic poisons. With a catalyst, a reaction which is kinetically inhibited by a loftier activation energy can take place in circumvention of this activation energy.
Heterogeneous catalysts are usually solids, powdered in guild to maximize their surface surface area. Of item importance in heterogeneous catalysis are the platinum group metals and other transition metals, which are used in hydrogenations, catalytic reforming and in the synthesis of commodity chemicals such as nitric acid and ammonia. Acids are an example of a homogeneous catalyst, they increase the nucleophilicity of carbonyls, allowing a reaction that would not otherwise proceed with electrophiles. The advantage of homogeneous catalysts is the ease of mixing them with the reactants, but they may also exist difficult to split from the products. Therefore, heterogeneous catalysts are preferred in many industrial processes.[39]
Reactions in organic chemical science
In organic chemistry, in addition to oxidation, reduction or acrid–base reactions, a number of other reactions can take place which involve covalent bonds between carbon atoms or carbon and heteroatoms (such as oxygen, nitrogen, halogens, etc.). Many specific reactions in organic chemistry are proper name reactions designated after their discoverers.
Substitution
In a commutation reaction, a functional group in a particular chemic compound is replaced by another group.[40] These reactions can be distinguished by the blazon of substituting species into a nucleophilic, electrophilic or radical substitution.

SN1 machinery
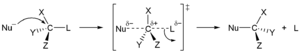
Due southNorthwardii machinery
In the outset type, a nucleophile, an cantlet or molecule with an backlog of electrons and thus a negative accuse or partial charge, replaces another cantlet or part of the "substrate" molecule. The electron pair from the nucleophile attacks the substrate forming a new bond, while the leaving grouping departs with an electron pair. The nucleophile may be electrically neutral or negatively charged, whereas the substrate is typically neutral or positively charged. Examples of nucleophiles are hydroxide ion, alkoxides, amines and halides. This blazon of reaction is plant mainly in aliphatic hydrocarbons, and rarely in effluvious hydrocarbon. The latter have high electron density and enter nucleophilic effluvious substitution simply with very strong electron withdrawing groups. Nucleophilic exchange can take place by ii different mechanisms, SNorthward1 and SNorth2. In their names, South stands for substitution, Northward for nucleophilic, and the number represents the kinetic order of the reaction, unimolecular or bimolecular.[41]

The 3 steps of an SouthN2 reaction. The nucleophile is dark-green and the leaving grouping is red
SNii reaction causes stereo inversion (Walden inversion)
The SN1 reaction proceeds in two steps. Get-go, the leaving group is eliminated creating a carbocation. This is followed past a rapid reaction with the nucleophile.[42]
In the SN2 mechanism, the nucleophile forms a transition country with the attacked molecule, and simply and so the leaving group is cleaved. These two mechanisms differ in the stereochemistry of the products. SN1 leads to the non-stereospecific addition and does not result in a chiral center, but rather in a set of geometric isomers (cis/trans). In dissimilarity, a reversal (Walden inversion) of the previously existing stereochemistry is observed in the SNorthwardtwo mechanism.[43]
Electrophilic exchange is the counterpart of the nucleophilic substitution in that the attacking atom or molecule, an electrophile, has low electron density and thus a positive charge. Typical electrophiles are the carbon atom of carbonyl groups, carbocations or sulfur or nitronium cations. This reaction takes place almost exclusively in aromatic hydrocarbons, where it is called electrophilic aromatic substitution. The electrophile assault results in the and so-chosen σ-complex, a transition land in which the aromatic organisation is abolished. And so, the leaving grouping, ordinarily a proton, is split off and the aromaticity is restored. An culling to aromatic substitution is electrophilic aliphatic substitution. Information technology is similar to the nucleophilic aliphatic substitution and also has two major types, SEastone and SouthwardEastii[44]

Mechanism of electrophilic aromatic substitution
In the third type of substitution reaction, radical substitution, the attacking particle is a radical.[40] This process normally takes the form of a chain reaction, for case in the reaction of alkanes with halogens. In the outset stride, light or heat disintegrates the halogen-containing molecules producing the radicals. So the reaction proceeds as an avalanche until two radicals see and recombine.[45]
-
- Reactions during the chain reaction of radical substitution


Addition and elimination
The addition and its counterpart, the emptying, are reactions which change the number of substituents on the carbon atom, and grade or cleave multiple bonds. Double and triple bonds can be produced by eliminating a suitable leaving grouping. Like to the nucleophilic exchange, in that location are several possible reaction mechanisms which are named after the respective reaction society. In the E1 machinery, the leaving group is ejected showtime, forming a carbocation. The next step, formation of the double bond, takes place with elimination of a proton (deprotonation). The leaving order is reversed in the E1cb mechanism, that is the proton is carve up off first. This machinery requires participation of a base.[46] Considering of the similar atmospheric condition, both reactions in the E1 or E1cb elimination e'er compete with the SNone substitution.[47]

E1 emptying

E1cb elimination

The E2 machinery too requires a base of operations, simply there the set on of the base and the elimination of the leaving group keep simultaneously and produce no ionic intermediate. In dissimilarity to the E1 eliminations, different stereochemical configurations are possible for the reaction product in the E2 machinery, because the attack of the base preferentially occurs in the anti-position with respect to the leaving group. Because of the similar conditions and reagents, the E2 elimination is always in competition with the SNii-exchange.[48]
![]()
Electrophilic addition of hydrogen bromide
The counterpart of elimination is the addition where double or triple bonds are converted into unmarried bonds. Similar to the substitution reactions, there are several types of additions distinguished past the type of the attacking particle. For instance, in the electrophilic addition of hydrogen bromide, an electrophile (proton) attacks the double bond forming a carbocation, which then reacts with the nucleophile (bromine). The carbocation can exist formed on either side of the double bond depending on the groups fastened to its ends, and the preferred configuration can be predicted with the Markovnikov's rule.[49] This rule states that "In the heterolytic addition of a polar molecule to an alkene or alkyne, the more than electronegative (nucleophilic) atom (or part) of the polar molecule becomes attached to the carbon atom bearing the smaller number of hydrogen atoms."[50]
If the addition of a functional group takes identify at the less substituted carbon atom of the double bail, then the electrophilic commutation with acids is not possible. In this case, i has to use the hydroboration–oxidation reaction, where in the commencement step, the boron atom acts as electrophile and adds to the less substituted carbon atom. At the 2nd step, the nucleophilic hydroperoxide or halogen anion attacks the boron atom.[51]
While the addition to the electron-rich alkenes and alkynes is mainly electrophilic, the nucleophilic addition plays an important role for the carbon-heteroatom multiple bonds, and especially its most of import representative, the carbonyl group. This procedure is ofttimes associated with an elimination, so that later on the reaction the carbonyl grouping is present over again. It is therefore called add-on-elimination reaction and may occur in carboxylic acid derivatives such every bit chlorides, esters or anhydrides. This reaction is oftentimes catalyzed by acids or bases, where the acids increase by the electrophilicity of the carbonyl group past binding to the oxygen atom, whereas the bases heighten the nucleophilicity of the attacking nucleophile.[52]
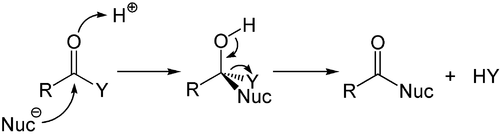
Acid-catalyzed improver-elimination mechanism
Nucleophilic addition of a carbanion or another nucleophile to the double bond of an alpha, beta unsaturated carbonyl chemical compound can proceed via the Michael reaction, which belongs to the larger class of cohabit additions. This is i of the well-nigh useful methods for the balmy formation of C–C bonds.[53] [54] [55]
Some additions which can not be executed with nucleophiles and electrophiles, can exist succeeded with free radicals. As with the free-radical exchange, the radical addition gain as a chain reaction, and such reactions are the basis of the free-radical polymerization.[56]
Other organic reaction mechanisms
![]()
The Cope rearrangement of 3-methyl-1,5-hexadiene

Mechanism of a Diels-Alder reaction

Orbital overlap in a Diels-Alder reaction
In a rearrangement reaction, the carbon skeleton of a molecule is rearranged to give a structural isomer of the original molecule. These include hydride shift reactions such as the Wagner-Meerwein rearrangement, where a hydrogen, alkyl or aryl group migrates from one carbon to a neighboring carbon. Most rearrangements are associated with the breaking and formation of new carbon-carbon bonds. Other examples are sigmatropic reaction such as the Cope rearrangement.[57]
Cyclic rearrangements include cycloadditions and, more than more often than not, pericyclic reactions, wherein two or more double bond-containing molecules form a cyclic molecule. An important instance of cycloaddition reaction is the Diels–Alder reaction (the and so-called [four+2] cycloaddition) betwixt a conjugated diene and a substituted alkene to form a substituted cyclohexene organization.[58]
Whether a certain cycloaddition would proceed depends on the electronic orbitals of the participating species, as only orbitals with the same sign of moving ridge office volition overlap and interact constructively to form new bonds. Cycloaddition is usually assisted by light or rut. These perturbations result in unlike organisation of electrons in the excited country of the involved molecules and therefore in different effects. For instance, the [4+2] Diels-Alder reactions can be assisted by heat whereas the [two+2] cycloaddition is selectively induced by light.[59] Considering of the orbital character, the potential for developing stereoisomeric products upon cycloaddition is limited, as described by the Woodward–Hoffmann rules.[60]
Biochemical reactions
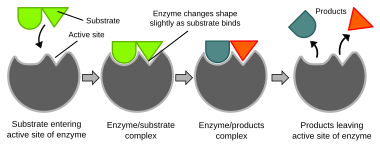
Illustration of the induced fit model of enzyme activeness
Biochemical reactions are mainly controlled by enzymes. These proteins can specifically catalyze a single reaction, so that reactions tin can exist controlled very precisely. The reaction takes identify in the agile site, a small part of the enzyme which is unremarkably found in a cleft or pocket lined by amino acid residues, and the residue of the enzyme is used mainly for stabilization. The catalytic action of enzymes relies on several mechanisms including the molecular shape ("induced fit"), bond strain, proximity and orientation of molecules relative to the enzyme, proton donation or withdrawal (acid/base of operations catalysis), electrostatic interactions and many others.[61]
The biochemical reactions that occur in living organisms are collectively known as metabolism. Among the about important of its mechanisms is the anabolism, in which dissimilar DNA and enzyme-controlled processes result in the production of large molecules such equally proteins and carbohydrates from smaller units.[62] Bioenergetics studies the sources of free energy for such reactions. Of import energy sources are glucose and oxygen, which can be produced by plants via photosynthesis or alloyed from food and air, respectively. All organisms use this energy to produce adenosine triphosphate (ATP), which can then be used to energize other reactions.
Applications

Thermite reaction proceeding in railway welding. Soon later on this, the liquid iron flows into the mould effectually the track gap.
Chemical reactions are central to chemical technology, where they are used for the synthesis of new compounds from natural raw materials such every bit petroleum, mineral ores, and oxygen in air. It is essential to make the reaction as efficient as possible, maximizing the yield and minimizing the amount of reagents, free energy inputs and waste material. Catalysts are especially helpful for reducing the energy required for the reaction and increasing its reaction rate.[63] [64]
Some specific reactions accept their niche applications. For example, the thermite reaction is used to generate light and heat in pyrotechnics and welding. Although information technology is less controllable than the more conventional oxy-fuel welding, arc welding and wink welding, it requires much less equipment and is still used to mend rails, particularly in remote areas.[65]
Monitoring
Mechanisms of monitoring chemic reactions depend strongly on the reaction charge per unit. Relatively dull processes tin be analyzed in situ for the concentrations and identities of the individual ingredients. Of import tools of real fourth dimension analysis are the measurement of pH and analysis of optical absorption (colour) and emission spectra. A less accessible but rather efficient method is introduction of a radioactive isotope into the reaction and monitoring how information technology changes over time and where it moves to; this method is ofttimes used to analyze redistribution of substances in the human being body. Faster reactions are usually studied with ultrafast laser spectroscopy where utilization of femtosecond lasers allows short-lived transition states to be monitored at fourth dimension scaled down to a few femtoseconds.[66]
Run across too
- Chemic equation
- Chemical reaction
- Substrate
- Reagent
- Catalyst
- Product
- Chemical reaction model
- Chemist
- Chemistry
- Combustion
- Limiting reagent
- List of organic reactions
- Mass balance
- Microscopic reversibility
- Organic reaction
- Reaction progress kinetic assay
- Reversible reaction
References
- ^ IUPAC, Compendium of Chemic Terminology, 2nd ed. (the "Gilded Volume") (1997). Online corrected version: (2006–) "chemical reaction". doi:10.1351/goldbook.C01033
- ^ Weyer, J. (1973). "Neuere Interpretationsmöglichkeiten der Alchemie". Chemie in unserer Zeit. seven (6): 177–181. doi:10.1002/ciuz.19730070604.
- ^ See Newman, William R. (2004). Promethean Ambitions: Alchemy and the Quest to Perfect Nature. Chicago: University of Chicago Press. ISBN9780226575247.
- ^ Kraus, Paul (1942–1943). Jâbir ibn Hayyân: Contribution à l'histoire des idées scientifiques dans l'Islam. I. Le corpus des écrits jâbiriens. 2. Jâbir et la scientific discipline grecque. Cairo: Institut Français d'Archéologie Orientale. ISBN9783487091150. OCLC 468740510. , vol. II, pp. 41–42.
- ^ Karpenko, Vladimír; Norris, John A. (2002). "Vitriol in the History of Chemistry". Chemické listy. 96 (12): 997–1005.
- ^ Friedman, Leonard J.; Friedman, Samantha J. (2008). The History of the Contact Sulfuric Acid Process (PDF). Boca Raton, Florida: Acid Engineering science & Consulting, Inc.
- ^ Stranges, Anthony Northward. (2000). "Germany'south synthetic fuel industry, 1935–1940". In Lesch, John East. (ed.). The German Chemic Industry in the Twentieth Century. Kluwer Academic Publishers. p. 170. ISBN978-0-7923-6487-0.
- ^ Brock, pp. 34–55
- ^ Brock, pp. 104–107
- ^ Myers, Richard (2009). The Basics of Chemistry. Greenwood Publishing Grouping. p. 55. ISBN978-0-313-31664-vii.
- ^ IUPAC, Compendium of Chemical Terminology, 2d ed. (the "Gold Book") (1997). Online corrected version: (2006–) "chemical reaction equation". doi:10.1351/goldbook.C01034
- ^ Corey, E.J. (1988). "Robert Robinson Lecture. Retrosynthetic thinking?essentials and examples". Chemical Gild Reviews. 17: 111–133. doi:10.1039/CS9881700111.
- ^ IUPAC, Compendium of Chemic Terminology, second ed. (the "Gilded Book") (1997). Online corrected version: (2006–) "elementary reaction". doi:ten.1351/goldbook.E02035
- ^ Frenking, Gernot (2006). "Elementarreaktionen". Römpp Chemie-Lexikon. Thieme.
- ^ a b Kandori, Hideki (2006). "Retinal Binding Proteins". In Dugave, Christophe (ed.). Cis-trans Isomerization in Biochemistry. Wiley-VCH. p. 56. ISBN978-3-527-31304-4.
- ^ Atkins, p. 114.
- ^ Atkins, pp. 106–108
- ^ a b Reaction Web
- ^ Atkins, p. 150
- ^ Atkins, p. 963
- ^ a b c d To react or not to react? Archived 2015-01-x at the Wayback Auto Utah Land Office of Educational activity. Retrieved four June 2011.
- ^ a b The half-dozen types of reaction – The Cavalcade o' Chemical science. Retrieved 11 February 2016
- ^ Wilbraham, Matta, Waterman, Stanley, Antony, Michael, Edward, Dennis (2012). Chemistry. Pearson. pp. 734–735. ISBN978-0-xiii-322662-1.
{{cite book}}: CS1 maint: multiple names: authors list (link) - ^ Glusker, Jenny P. (1991). "Structural Aspects of Metal Liganding to Functional Groups in Proteins". In Christian B. Anfinsen (ed.). Advances in Poly peptide Chemistry. Vol. 42. San Diego: Academic Press. p. 7. ISBN978-0-12-034242-6.
- ^ Guo, Liang-Hong; Allen, H.; Hill, O. (1991). "Direct Electrochemistry of Proteins and Enzymes". In A.K. Sykes (ed.). Advances in Inorganic Chemistry. Vol. 36. San Diego: Academic Printing. p. 359. ISBN978-0-12-023636-7.
- ^ Wiberg, pp. 289–290
- ^ Wiberg, p. 409
- ^ Wiberg, pp. 1180–1205
- ^ IUPAC, Compendium of Chemical Terminology, 2nd ed. (the "Gold Book") (1997). Online corrected version: (2006–) "conjugate acid–base pair". doi:10.1351/goldbook.C01266
- ^ IUPAC, Compendium of Chemical Terminology, 2nd ed. (the "Gold Book") (1997). Online corrected version: (2006–) "precipitation". doi:10.1351/goldbook.P04795
- ^ Wingender, Jörg; Ortanderl, Stefanie (July 2009). "Ausfällung". Römpp Chemie-Lexikon. Thieme.
- ^ Meyer, H. Jürgen (2007). "Festkörperchemie". In Erwin Riedel (ed.). Modern Inorganic Chemistry (in German language) (3rd ed.). de Gruyter. p. 171. ISBN978-3-eleven-019060-i.
- ^ Wintterlin, J. (1997). "Atomic and Macroscopic Reaction Rates of a Surface-Catalyzed Reaction". Science. 278 (5345): 1931–4. Bibcode:1997Sci...278.1931W. doi:10.1126/science.278.5345.1931. PMID 9395392.
- ^ Waldmann, T.; Künzel, D.; Hoster, H.E.; Groß, A.; Behm, R.J.R. (2012). "Oxidation of an Organic Adlayer: A Bird's Eye View". Periodical of the American Chemical Club. 134 (21): 8817–8822. doi:ten.1021/ja302593v. PMID 22571820.
- ^ Atkins, pp. 937–950
- ^ Saunders, David Stanley (2002). Insect clocks (Third ed.). Amsterdam: Elsevier. p. 179. ISBN978-0-444-50407-4.
- ^ IUPAC, Compendium of Chemic Terminology, 2nd ed. (the "Gold Book") (1997). Online corrected version: (2006–) "goad". doi:10.1351/goldbook.C00876
- ^ IUPAC, Compendium of Chemical Terminology, 2d ed. (the "Gold Volume") (1997). Online corrected version: (2006–) "inhibitor". doi:x.1351/goldbook.I03035
- ^ Elschenbroich, Christoph (2008). Organometallchemie (6th ed.). Wiesbaden: Vieweg+Teubner Verlag. p. 263. ISBN978-3-8351-0167-viii.
- ^ a b March, Jerry (1985), Avant-garde Organic Chemical science: Reactions, Mechanisms, and Structure (3rd ed.), New York: Wiley, ISBN0-471-85472-7
- ^ Hartshorn, South.R. (1973). Aliphatic Nucleophilic Commutation. London: Cambridge University Printing. p. 1. ISBN978-0-521-09801-4.
- ^ Bateman, Leslie C.; Church, Mervyn One thousand.; Hughes, Edward D.; Ingold, Christopher K.; Taher, Nazeer Ahmed (1940). "188. Machinery of substitution at a saturated carbon cantlet. Part XXIII. A kinetic demonstration of the unimolecular solvolysis of alkyl halides. (Department E) a general discussion". Journal of the Chemic Gild: 979. doi:10.1039/JR9400000979.
- ^ Brückner, pp. 63–77
- ^ Brückner, pp. 203–206
- ^ Brückner, p. sixteen
- ^ Brückner, p. 192
- ^ Brückner, p. 183
- ^ Brückner, p. 172
- ^ Wiberg, pp. 950, 1602
- ^ IUPAC, Compendium of Chemical Terminology, second ed. (the "Gold Book") (1997). Online corrected version: (2006–) "Markownikoff rule". doi:10.1351/goldbook.M03707
- ^ Brückner, p. 125
- ^ Latscha, Hans Peter; Kazmaier, Uli; Klein, Helmut Alfons (2008). Organische Chemie: Chemie-basiswissen II (in German language). Vol. 2 (6th ed.). Springer. p. 273. ISBN978-3-540-77106-vii.
- ^ Organic Reactions. 2004. doi:10.1002/0471264180. ISBN978-0-471-26418-seven.
- ^ Hunt, Ian. "Chapter eighteen: Enols and Enolates — The Michael Addition reaction". Academy of Calgary.
- ^ Brückner, p. 580
- ^ Lechner, Manfred; Gehrke, Klaus; Nordmeier, Eckhard (2003). Macromolecular Chemistry (3rd ed.). Basel: Birkhäuser. pp. 53–65. ISBN978-three-7643-6952-1.
- ^ Pull a fast one on, Marye Anne; Whitesell, James K. (2004). Organic chemistry (Third ed.). Jones & Bartlett. p. 699. ISBN978-0-7637-2197-8.
- ^ Diels, O.; Alder, K. (1928). "Synthesen in der hydroaromatischen Reihe". Justus Liebig's Annalen der Chemie. 460: 98–122. doi:x.1002/jlac.19284600106.
- ^ Brückner, pp. 637–647
- ^ Woodward, R.B.; Hoffmann, R. (1965). "Stereochemistry of Electrocyclic Reactions". Periodical of the American Chemic Society. 87 (2): 395–397. doi:x.1021/ja01080a054.
- ^ Karlson, Peter; Doenecke, Detlef; Koolman, Jan; Fuchs, Georg; Gerok, Wolfgang (2005). Karlson Biochemistry and Pathobiochemistry (in German) (16th ed.). Thieme. pp. 55–56. ISBN978-3-13-357815-viii.
- ^ IUPAC, Compendium of Chemic Terminology, 2nd ed. (the "Gold Book") (1997). Online corrected version: (2006–) "anabolism". doi:ten.1351/goldbook.A00314
- ^ Emig, Gerhard; Klemm, Elias (2005). Technical Chemistry (in German) (fifth ed.). Springer. pp. 33–34. ISBN978-3-540-23452-4.
- ^ Trost, B. (1991). "The cantlet economy – a search for synthetic efficiency". Science. 254 (5037): 1471–1477. Bibcode:1991Sci...254.1471T. doi:x.1126/science.1962206. PMID 1962206.
- ^ Weismantel, Guy E (1999). John J. McKetta (ed.). Encyclopedia of Chemic Processing and Design. Vol. 67. CRC Press. p. 109. ISBN978-0-8247-2618-8 https://books.google.com/books?id=MfjDlUe8Kc0C&pg=PA109.
- ^ Atkins, p. 987
Bibliography
- Atkins, Peter Due west.; Julio de Paula (2006). Physical Chemistry (4th ed.). Weinheim: Wiley-VCH. ISBN978-three-527-31546-8.
- Brock, William H. (1997). Viewegs Geschichte der Chemie (in German). Braunschweig: Vieweg. ISBN978-three-540-67033-9.
- Brückner, Reinhard (2004). Reaktionsmechanismen (in High german) (third ed.). München: Spektrum Akademischer Verlag. ISBN978-3-8274-1579-0.
- Wiberg, Egon, Wiberg, Nils and Holleman, Arnold Frederick (2001). Inorganic chemistry. Bookish Press. ISBN978-0-12-352651-9.
{{cite book}}: CS1 maint: multiple names: authors list (link) - . Encyclopædia Britannica. Vol. half dozen (11th ed.). 1911. pp. 26–33.

A Certain Chemical Reaction Releases,
Source: https://en.wikipedia.org/wiki/Chemical_reaction
Posted by: mccollisteraloortat.blogspot.com
0 Response to "A Certain Chemical Reaction Releases"
Post a Comment